



RT-qPCR은 일반적인 PCR 기술에서 개발되었습니다.기존의 PCR 반응 시스템에 형광 화학물질(형광 염료 또는 형광 프로브)을 추가하고 서로 다른 발광 메커니즘에 따라 PCR annealing 및 extension 과정을 실시간으로 감지합니다.매체의 형광 신호 변화는 PCR의 각 주기에서 생성물 변화량을 계산하는 데 사용됩니다.현재 가장 보편적인 방법은 형광염료법과 프로브법이다.
형광 염료 방법:
SYBR Green Ⅰ, PicoGreen, BEBO 등 일부 형광염료는 스스로 발광하지 않고 dsDNA의 minor groove에 결합한 후 형광을 발한다.따라서 PCR 반응 초기에는 기계가 형광 신호를 감지할 수 없습니다.반응이 annealing-extension(two-step method) 또는 extension stage(three-step method)로 진행되면 이때 이중가닥이 열리고 새로운 DNA polymerase가 가닥을 합성하는 동안 형광 분자가 dsDNA 마이너 그루브(minor groove)에 결합하여 형광을 발한다.PCR 사이클의 수가 증가함에 따라 점점 더 많은 염료가 dsDNA와 결합하고 형광 신호도 지속적으로 향상됩니다.SYBR Green Ⅰ을 예로 들어 보겠습니다.
프로브 방법:
Taqman 프로브는 가장 일반적으로 사용되는 가수분해 프로브입니다.프로브의 5' 말단에는 일반적으로 FAM인 형광 그룹이 있습니다.프로브 자체는 표적 유전자에 상보적인 서열입니다.fluorophore의 3' 끝에 형광 quenching 그룹이 있습니다.형광 공명 에너지 전달(Förster resonance energy transfer, FRET) 원리에 따르면, 리포터 형광기(공여체 형광 분자)와 소광 형광기(억셉터 형광 분자)가 여기 스펙트럼이 중첩되어 거리가 매우 가까워지면(7-10nm) 도너 분자의 여기가 억셉터 분자의 형광을 유도할 수 있는 반면, 자가형광은 약해진다.따라서 PCR 반응 초기에 프로브가 시스템에서 자유롭고 온전할 때 리포터 형광 그룹은 형광을 방출하지 않습니다.어닐링 시 프라이머와 프로브가 템플릿에 결합합니다.연장 단계에서 중합효소는 지속적으로 새로운 사슬을 합성합니다.DNA 중합효소는 5'-3' 엑소뉴클레아제 활성을 가지고 있습니다.프로브에 도달하면 DNA 폴리머라제가 템플릿에서 프로브를 가수분해하고 리포터 형광 그룹을 소광제 형광 그룹에서 분리하고 형광 신호를 방출합니다.탐침과 주형은 일대일 관계이기 때문에 탐침법이 염색법보다 검사의 정확도와 민감도 면에서 우수하다.
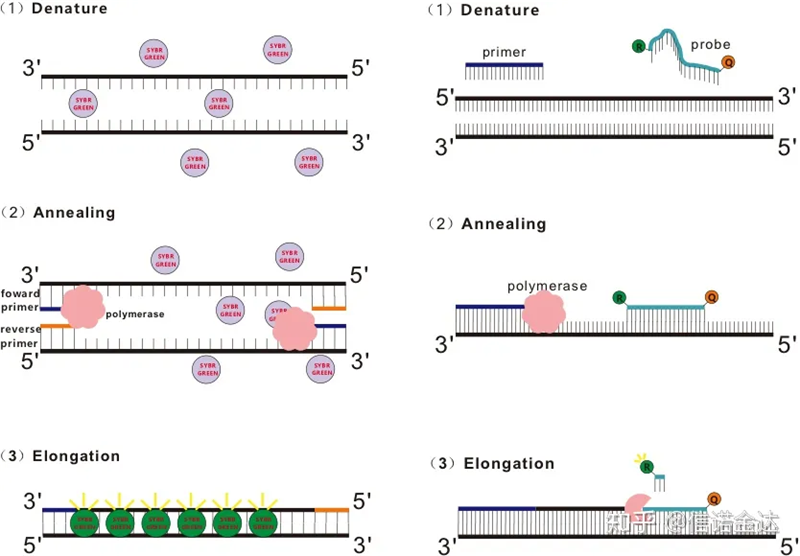
그림 1 qRT-PCR의 원리
프라이머 디자인
원칙:
프라이머는 핵산 시리즈의 보존된 영역에 설계되어야 하며 특이성을 가져야 합니다.
cDNA 염기서열을 사용하는 것이 가장 좋으며 mRNA 염기서열도 가능하다.그렇지 않은 경우 DNA 서열의 cds 영역 디자인을 찾으십시오.
형광 정량 산물의 길이는 80-150bp, 가장 긴 것은 300bp, 프라이머 길이는 일반적으로 17-25 염기 사이이며, 업스트림과 다운스트림 프라이머의 차이가 너무 크지 않아야 합니다.
G+C 함량은 40~60% 사이이며 45~55%가 가장 좋다.
TM 값은 58-62도 사이입니다.
primer dimer와 self-dimer를 피하고, (연속된 상보적인 염기가 4쌍 이상 나타나지 않도록) 헤어핀 구조, 불가피한 경우 ΔG<4.5kJ/mol*로 만드십시오. 역전사 과정에서 gDNA가 제거되었는지 확인할 수 없다면 Clean, intron *3' 말단은 변형될 수 없도록 설계하는 것이 가장 좋으며, AT, GC rich region을 피하기 위해서는 T/C, A/G 연속 구조(2-3) primer 및 non-
특이적 이질적으로 증폭된 서열의 상동성은 바람직하게는 70% 미만이거나 8개의 상보적인 염기 상동성을 갖는다.
데이터 베이스:
CottonFGD 키워드로 검색
프라이머 디자인:
IDT-qPCR 프라이머 디자인
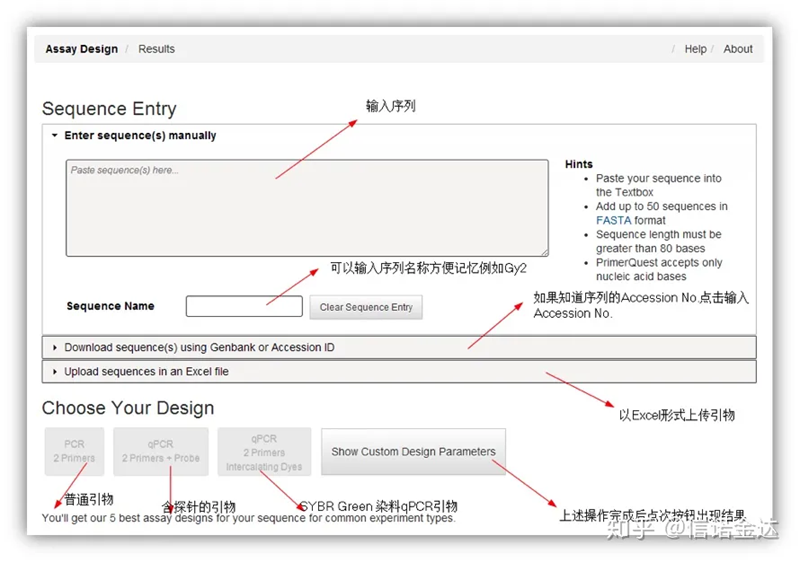
Fig2 IDT 온라인 프라이머 디자인 도구 페이지
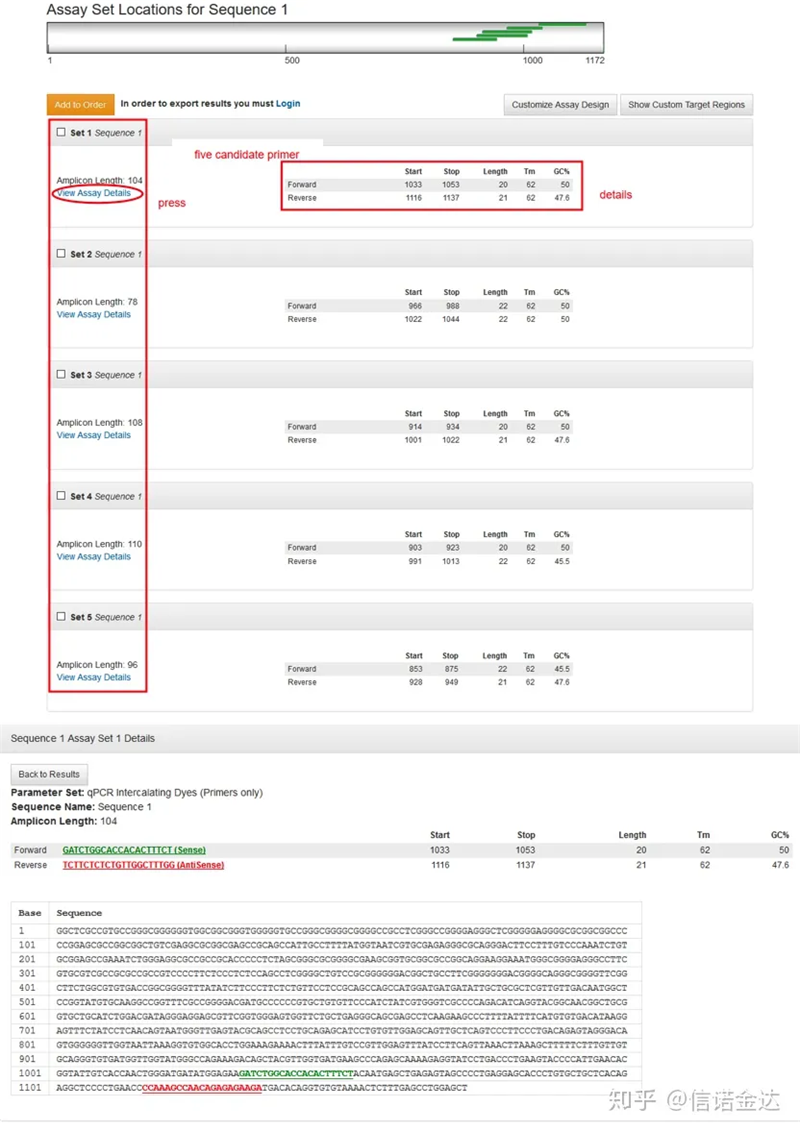
Fig3 결과 페이지 표시
lncRNA 프라이머 설계:
lncRNA:mRNA와 동일한 단계.
miRNA:스템-루프 방법의 원리: 모든 miRNA는 약 23nt의 짧은 염기서열이기 때문에 직접적인 PCR 검출이 불가능하므로 스템-루프 염기서열 도구를 사용합니다.스템-루프 서열은 약 50nt의 단일가닥 DNA로서, 스스로 헤어핀 구조를 형성할 수 있다.3' 말단은 miRNA 부분 단편에 상보적인 서열로 설계될 수 있으며, 그 다음 표적 miRNA는 역전사 동안 스템-루프 서열에 연결될 수 있고, 총 길이는 70bp에 도달할 수 있으며, 이는 qPCR에 의해 결정된 증폭된 산물의 길이와 일치한다.테일링 miRNA 프라이머 디자인 .
증폭 관련 감지:
온라인 폭발 데이터베이스: 시퀀스 유사성에 의한 CottonFGD 폭발
로컬 폭발: Blast+를 사용하여 로컬 폭발을 수행하는 것을 참조하십시오. Linux 및 macos는 로컬 데이터베이스를 직접 설정할 수 있으며, win10 시스템은 ubuntu bash를 설치한 후 수행할 수도 있습니다.로컬 폭발 데이터베이스 및 로컬 폭발 생성 ;win10에서 우분투 bash를 엽니다.
참고: 고지대 목화와 바다 섬 목화는 사배체 작물이므로 도열의 결과는 종종 두 개 이상의 성냥이 될 것입니다.과거에 폭발을 수행하기 위해 NAU cds를 데이터베이스로 사용하면 SNP 차이가 거의 없는 두 개의 상동 유전자를 찾을 가능성이 높습니다.보통 두 상동 유전자는 프라이머 디자인으로 분리할 수 없기 때문에 같은 것으로 취급한다.명백한 인델이 있는 경우 보통 인델 위에 프라이머를 디자인하는데, 이는 프라이머의 2차 구조로 이어질 수 있어 자유에너지가 높아져 증폭 효율 저하로 이어지지만 이는 어쩔 수 없는 현상이다.
프라이머 2차 구조의 검출:
단계:올리고 7 열기 → 템플릿 시퀀스 입력 → 하위 창 닫기 → 저장 → 템플릿에서 프라이머 위치, ctrl+D를 눌러 프라이머 길이 설정 → 다양한 이차 구조 분석(예: 자기 이합체, 헤테로다이머, 헤어핀, 미스매치 등) 그림 4의 마지막 두 그림은 프라이머의 테스트 결과입니다.프론트 프라이머의 결과는 양호하고 명백한 이합체 및 헤어핀 구조가 없으며 연속적인 상보적 염기가 없으며 자유 에너지의 절대값은 4.5 미만인 반면 후면 프라이머는 연속적인 6개의 염기가 상보적이며 자유 에너지는 8.8입니다.또한 3′말단에는 더 심각한 이량체가 나타나며 연속된 4개의 염기의 이량체가 나타난다.자유 에너지는 높지 않지만 3' 이합체 Chl은 증폭 특이성과 증폭 효율에 심각한 영향을 미칠 수 있습니다.또한 헤어핀, 이종이중체, 미스매치 등을 확인해야 한다.
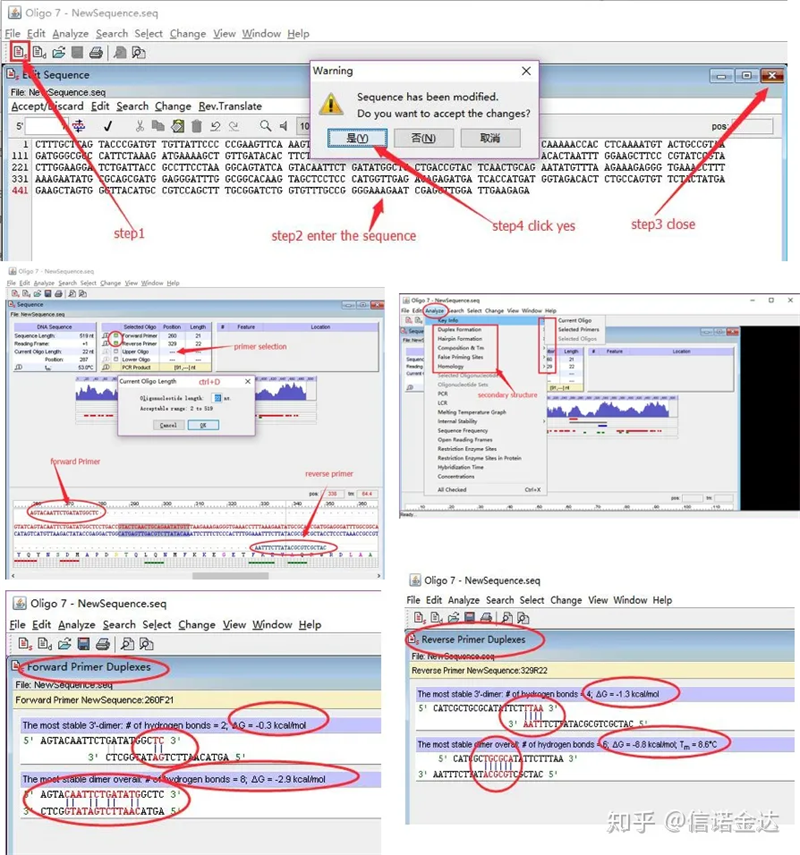
Fig3 oligo7 검출 결과
증폭 효율 감지:
PCR 반응의 증폭 효율은 PCR 결과에 심각한 영향을 미칩니다.또한 qRT-PCR에서 증폭 효율은 정량적 결과에 특히 중요합니다.반응 버퍼에서 다른 물질, 기계 및 프로토콜을 제거합니다.프라이머의 품질도 qRT-PCR의 증폭 효율에 큰 영향을 미칩니다.결과의 정확성을 보장하기 위해 상대 형광 정량과 절대 형광 정량 모두 프라이머의 증폭 효율을 감지해야 합니다.효과적인 qRT-PCR 증폭 효율은 85%에서 115% 사이인 것으로 알려져 있습니다.두 가지 방법이 있습니다.
1. 표준 곡선 방법:
ㅏ.혼합 cDNA
비.그라데이션 희석
c.qPCR
디.증폭 효율을 계산하기 위한 선형 회귀 방정식
2. LinRegPCR
LinRegPCR은 SYBR Green 또는 유사한 화학을 기반으로 정량적 PCR(qPCR) 데이터라고도 하는 실시간 RT-PCR 데이터 분석을 위한 프로그램입니다.이 프로그램은 기준선이 아닌 수정된 데이터를 사용하고, 각 샘플에 대해 기준선 수정을 개별적으로 수행하고, 선형성 창을 결정한 다음 선형 회귀 분석을 사용하여 PCR 데이터 세트를 통해 직선을 맞춥니다.이 선의 기울기에서 각 개별 샘플의 PCR 효율이 계산됩니다.앰플리콘당 평균 PCR 효율 및 샘플당 Ct 값은 임의의 형광 단위로 표현되는 샘플당 시작 농도를 계산하는 데 사용됩니다.데이터 입력 및 출력은 Excel 스프레드시트를 통해 이루어집니다.샘플만
혼합이 필요하며 그라데이션이 없습니다.
다음 단계가 필요합니다.(예를 들어 Bole CFX96을 예로 들어보겠습니다. 명확한 ABI가 있는 머신은 아닙니다.)
실험:표준 qPCR 실험입니다.
qPCR 데이터 출력:LinRegPCR은 두 가지 형태의 출력 파일을 인식할 수 있습니다: RDML 또는 정량화 증폭 결과.실제로 기계에 의한 사이클 수와 형광 신호의 실시간 검출값이며, 선형 세그먼트 효율의 형광 변화값을 분석하여 증폭도를 얻는다.
데이터 선택: 이론적으로 RDML 값은 사용 가능해야 합니다.내 컴퓨터의 문제는 소프트웨어가 RDML을 인식하지 못하는 것으로 추정되므로 Excel 출력 값을 원본 데이터로 가지고 있습니다.샘플 추가 실패 등 데이터에 대한 대략적인 스크리닝을 먼저 수행하는 것이 좋습니다. 포인트는 출력 데이터에서 삭제할 수 있습니다(물론 삭제할 수는 없습니다. LinRegPCR은 이후 단계에서 이 포인트를 무시합니다).
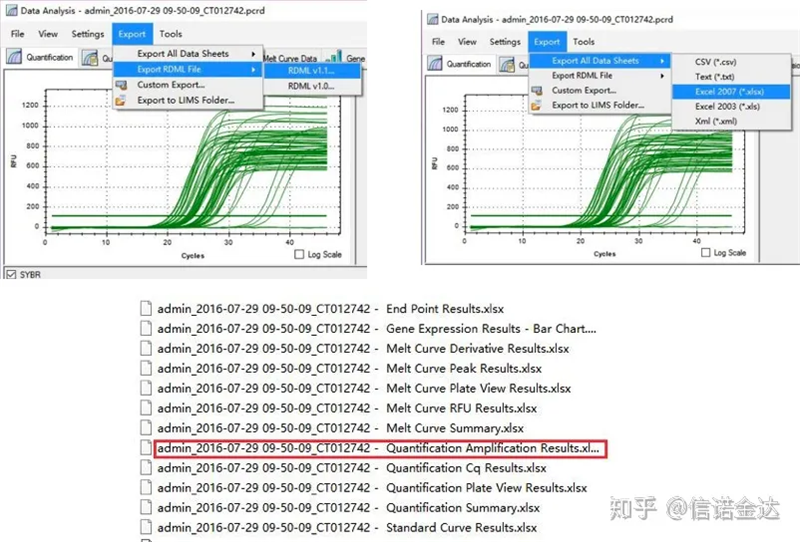
Fig5 qPCR 데이터 내보내기
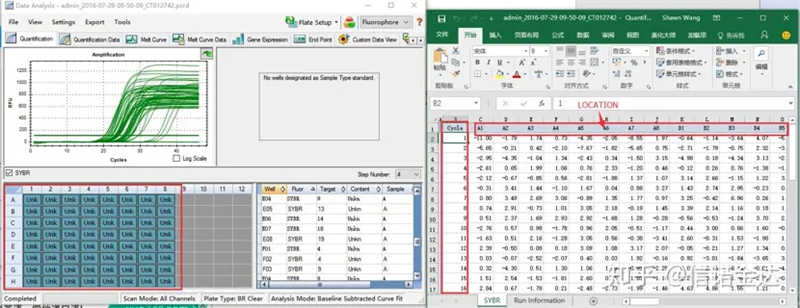
후보 샘플의 Fig6 선택
데이터 투입:Qualification amplification results.xls 열기, → LinRegPCR 열기 → 파일 열기 → Excel에서 읽기 → 그림 7과 같이 매개변수 선택 → 확인 → 기준선 결정 클릭
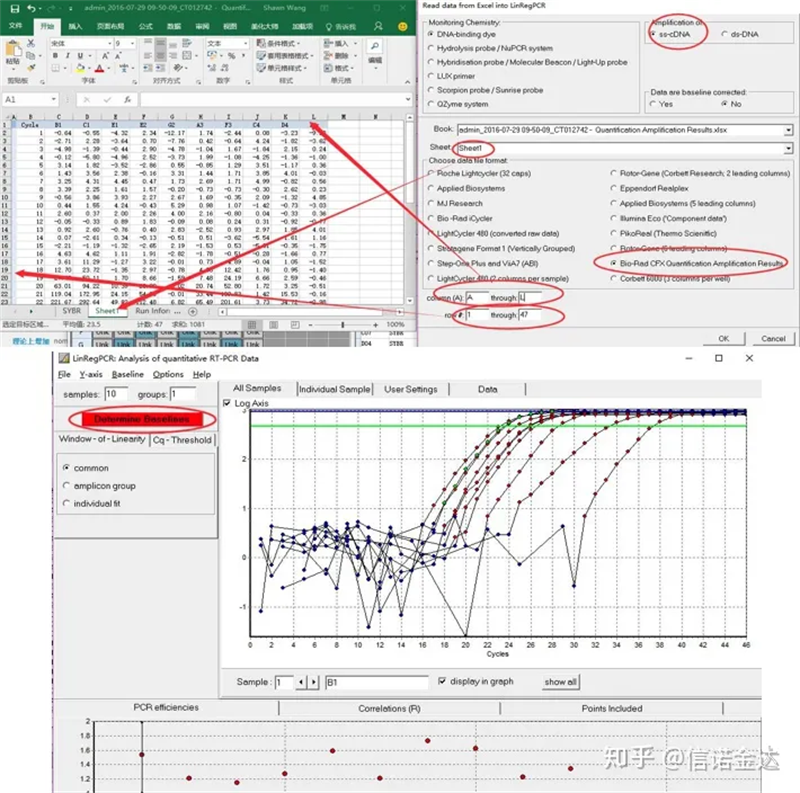
Fig7 linRegPCR 데이터 입력 단계
결과:반복이 없으면 그룹화가 필요하지 않습니다.반복되는 경우 샘플 그룹화에서 그룹화를 편집할 수 있으며 식별자에 유전자 이름을 입력하면 동일한 유전자가 자동으로 그룹화됩니다.마지막으로 파일을 클릭하고 Excel을 내보내고 결과를 봅니다.증폭 효율과 각 웰의 R2 결과가 표시됩니다.둘째, 그룹으로 나누면 보정된 평균 증폭 효율이 표시됩니다.각 프라이머의 증폭 효율이 85%에서 115% 사이인지 확인합니다.너무 크거나 작으면 프라이머의 증폭 효율이 나쁘다는 뜻이다.
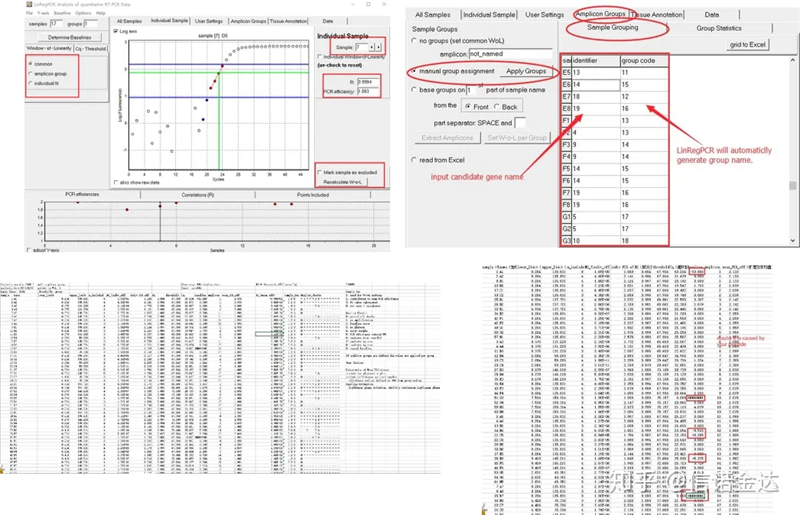
그림 8 결과 및 데이터 출력
실험 과정:
RNA 품질 요건:
청정:1.72.0은 잔류 이소티오시아네이트가 있을 수 있음을 나타냅니다.깨끗한 핵산 A260/A230은 약 2 이어야 합니다. 230 nm에서 강한 흡수가 있으면 페네이트 이온과 같은 유기 화합물이 있음을 나타냅니다.또한 1.5% agarose gel 전기영동으로 검출할 수 있다.ssRNA에는 denaturation이 없고 분자량 대수는 선형 관계가 없으며 분자량을 올바르게 표현할 수 없기 때문에 마커를 가리킵니다.농도: 이론적으로~ 아니다100ng/ul 이하, 농도가 너무 낮으면 일반적으로 순도가 낮고 높지 않음
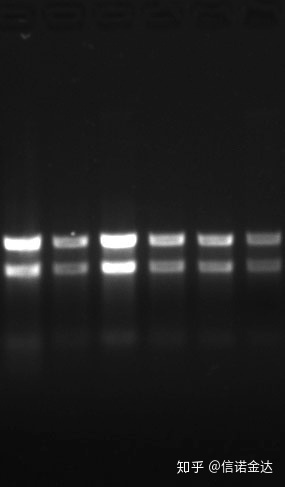
Fig9 RNA 겔
또한 시료가 귀하고 RNA농도가 높은 경우에는 추출 후 aliquot하고 RNA를 최종농도 100-300ng/ul로 희석하여 역전사를 하는 것을 권장합니다.~ 안에역전사의 과정, mRNA가 전사될 때 polyA tail에 특이적으로 결합할 수 있는 oligo(dt) 프라이머가 역전사에 사용되는 반면, lncRNA와 circRNA는 total RNA의 역전사에 random hexamer(Random 6 mer) 프라이머를 사용하고, miRNA의 경우 역전사에 miRNA-specific neck-loop primer가 사용된다.이제 많은 회사에서 특수 테일링 키트를 출시했습니다.스템-루프 방식의 경우 테일링 방식이 더 편리하고 처리량이 많으며 시약을 절약할 수 있지만, 같은 계열의 miRNA를 구별하는 효과는 스템-루프 방식만큼 좋지 않아야 합니다.각 역전사 키트에는 유전자 특이적 프라이머(스템 루프)의 농도에 대한 요구 사항이 있습니다.miRNA에 사용되는 내부 참조는 U6입니다.스템-루프 반전 과정에서 U6의 튜브를 별도로 반전시켜야 하며 U6의 전면 및 후면 프라이머를 직접 추가해야 합니다.circRNA와 lncRNA 모두 HKG를 내부 참조로 사용할 수 있습니다.~ 안에cDNA 검출,
RNA에 문제가 없다면 cDNA도 괜찮을 것입니다.그러나 실험의 완성도를 추구한다면 gDNA와 cds를 구별할 수 있는 내부 참조 유전자(Reference gene, RG)를 사용하는 것이 가장 좋다.일반적으로 RG는 하우스키핑 유전자입니다., HKG) 도 10에 도시된 바와 같이;당시 콩 저장 단백질을 만들고 있었는데 인트론이 포함된 액틴7을 내부 참조로 사용했습니다.이 프라이머의 증폭 단편의 gDNA 내 크기는 452bp였으며, cDNA를 주형으로 사용했을 경우 142bp였다.그 후 테스트 결과 cDNA의 일부가 gDNA에 의해 실제로 오염된 것으로 나타났으며 역전사 결과에도 문제가 없으며 PCR의 주형으로 사용할 수 있음을 증명했습니다.cDNA로 직접 agarose gel 전기영동을 하는 것은 소용이 없고, 확산 밴드(diffuse band)라 설득력이 없다.
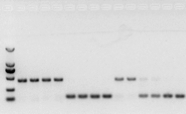
그림 10 cDNA 검출
qPCR 조건의 결정일반적으로 키트의 프로토콜에 따르면 주로 tm 값 단계에서 문제가 없습니다.Primer 설계 시 일부 primer가 제대로 설계되지 않아 tm 값과 이론적 60°C 사이에 큰 차이가 발생하는 경우 cDNA 샘플을 혼합한 후 Primer를 사용하여 Gradient PCR을 실행하고 Band가 없는 온도를 TM 값으로 설정하지 않도록 합니다.
데이터 분석
종래의 상대 형광 정량적 PCR 처리 방법은 기본적으로 2에 따른다.-ΔΔCT.데이터 처리 템플릿.
관련 상품:
RT Easy I (First strand cDNA 합성을 위한 Master Premix)
RT Easy II(qPCR을 위한 첫 가닥 cDNA 합성을 위한 Master Premix)
게시 시간: 2023년 3월 14일



